Example Experiment: Detecting p38 Activation in HeLa Cells Following Anisomycin Treatment
Background
Accurate quantification of protein expression and/or post-translational modifications is important for advancing both basic and translational research. Given the flexibility, reproducibility, and higher throughput of the In‑Cell Western™ Assay, it offers a convenient alternative to Western blotting and is a powerful platform for meaningful in situ analyses. The In-Cell Western microplate format can be used to analyze:
- Protein phosphorylation and signaling (1 - 3)
- Off-target effects of drugs on signaling pathways (4)
- Timing and kinetics of signaling events (5, 6)
- Quantification of viral load (7 - 11)
- Genotoxicity assays (12, 13)
- Cell proliferation and apoptosis assays (14)
- Bacterial-induced epithelial signaling (15)
- Glycoprotein analysis (16, 17)
- Library screening (18 - 20)
- Screening of monoclonal antibody clones (21)
Introduction
Understanding the processes leading to programmed cell death (apoptosis) is of utmost importance in the study of many diseases, including autoimmune diseases, neurological diseases, and cancer (22). For example, damaged cells that do not enter the apoptotic pathway may continue to proliferate and become cancerous (23). Due to the importance and complexity of the apoptosis signaling pathway, it is not surprising that this pathway plays an important role in numerous diseases. Consequently, understanding how new therapies impact this complex pathway is of great importance to the drug discovery field.
In this experiment, we illustrate how the In-Cell Western Assay can be utilized to quantify the apoptotic response after drug treatment in a human cancer cell line. The p38 mitogen-activated protein kinases (MAPKs) are a family of important signaling molecules that control cellular responses to stress stimuli, such as various environmental stressors, inflammatory cytokines, and chemotherapeutic agents (24, 25). p38 MAPKs are activated through the dual phosphorylation of their Thr-Gly-Tyr motif and regulate many physiological responses including, cell growth, proliferation, differentiation, migration, and apoptosis (24).
Experimental Design
In the following example, p38 activation is monitored using an antibody that detects endogenous levels of phosphorylated (singular or dual) p38 MAPK. This antibody does not recognize p38 that has not been phosphorylated. Phosphorylated p-38 levels are then normalized against total ERK2. Anisomycin, a protein synthesis inhibitor, has been shown to be an activator of the p38 MAPK pathway and trigger apoptosis in several cancer cell lines (26 - 33). Therefore, we investigated the effects of anisomycin on apoptosis as measured by p38 activation in HeLa cells by In‑Cell Western™ Assay.
Required Reagents
LI‑COR Reagents
-
IRDye® 800CW Goat anti-Rabbit Secondary Antibody (LI-COR P/N 925-32211 or 926-32211)
-
IRDye 680RD Goat anti-Mouse Secondary Antibody (LI-COR P/N 925-68070 or 926-68070)
-
Intercept® (PBS) Blocking Buffer
Odyssey® Blocking Buffer was used in the original experiment and has been discontinued. Intercept Blocking Buffer is now available instead of Odyssey Blocking Buffer.
Additional Reagents
- 1X PBS wash buffer
- Standard tissue culture reagents (serum, DMEM media, trypsin, 1X PBS)
- HeLa cells (ATCC, P/N CCL-2)
- Black-sided 96-well or 384-well microplates with clear well bottoms
- Anisomycin (Sigma-Aldrich P/N A9789)
- Anti-phospho p38 antibody (Cell Signaling Technology, P/N 9211)
- Normalization antibody (e.g., anti-total ERK2 (Santa Cruz Biotechnology, P/N SC-1647))
- 20% Tween® 20
- 37% formaldehyde
- 10% Triton® X-100
Prepare Cells
-
Allow HeLa cell growth in a T75 flask using standard tissue culture procedures until ~80% confluency is achieved (~1.5 x 107 cells; DMEM, 10% FBS; Gibco®).
-
Remove growth media and wash cells with sterile 1X PBS (room temperature (RT)).
-
Add trypsin and incubate 3-5 minutes at 37 °C to displace cells.
-
Neutralize displaced cells with culture media and pellet by centrifugation (500 x g).
-
Remove supernatant and disrupt the cell pellet manually by hand-tapping the collection tube.
To maintain cell integrity, do not pipet or vortex during pellet disruption.
-
Resuspend cells in 20 mL of complete media and count cells using a hemocytometer.
-
Reconstitute and dilute cells with complete media to a concentration of 75,000 cells/mL.
-
Manually mix the cell suspension thoroughly.
-
Under sterile conditions, dispense 200 µL of the cell suspension per well in a 96-well plate (15,000 cells plated per well).
-
Incubate cells at 37 °C with 5% CO2 in air atmosphere. Monitor cell density until ~80% consistency is achieved.
Treat Cells
- Warm serum-free media (DMEM, Gibco) to 37 °C. In a fresh 96-well microplate, prepare two-fold serial dilutions of anisomycin, ranging from 2 to 1,000 nM (0.5 to 265 ng/mL). Leave the first and second wells without anisomycin (resting cells as control). See "Experimental Results".
- Add either serum-free media for resting cells (mock) or serum-free media containing dilution series (1:2) of anisomycin ranging in concentration from 0.07 - 40 µM. Add 100 µL of resting or activation media per well.
- Remove complete media from plate wells by aspiration or manual displacement.
- Transfer media and anisomycin dilutions from the dilution plate into the cell-containing plate.
- Incubate at 37 °C with 5% CO2 for 30 minutes.
Fix and Permeabilize Cells
Fix Cells
-
Prepare fresh Fixing Solution as follows:
1X PBS 45 mL 37% Formaldehyde 5 mL 3.7% Formaldehyde 50 mL -
When incubation period is complete, carefully remove activation media manually or by aspiration to avoid detaching the cells.
-
Using a multi-channel pipettor, add 150 µL of fresh Fixing Solution (RT). Add the solution by pipetting down the sides of the wells carefully to avoid detaching the cells from the well bottom.
-
Allow incubation on bench top for 20 minutes at RT with no shaking.
Permeabilize Cells
-
Prepare Triton® Washing Solution as follows:
1X PBS 495 mL 10% Triton X-100 5 mL 1X PBS + 0.1% Triton X-100 500 mL -
Remove Fixing Solution to an appropriate waste container (contains formaldehyde).
-
Using a multi-channel pipettor, add 200 µL Triton Washing Solution (RT). Add the solution down the sides of the wells carefully to avoid detaching the cells.
-
Allow plate to shake on a rotator for 5 minutes at RT.
-
Repeat washing steps 4 more times, removing wash manually each time.
Do not allow cells to become dry during washing. Immediately add the next wash after manual disposal.
Block Cells
- Using a multi-channel pipettor, add 150 µL of Intercept® Blocking Buffer to each well. Add the solution by pipetting down the sides of the wells carefully to avoid detaching the cells.
- Allow blocking for 1.5 hours at RT with moderate shaking on a plate shaker.
Primary Antibodies
Dilute Primary Antibodies
-
Dilute the two primary antibodies in Intercept Blocking Buffer. Combine the following solutions as defined below for phospho-p38 target analysis, using total ERK2 for normalization:
Phospho-p38 (rabbit; 1:100 dilution in the combined solution; Cell Signaling Technology, P/N 9211)
Total ERK2 (mouse; 1:100 dilution in the combined solution; Santa Cruz Biotechnology P/N SC-1647)
-
Mix the primary antibody solution thoroughly before adding to wells.
Incubate with Primary Antibodies
- Remove blocking buffer and add 50 µL of the desired primary antibody or antibodies in Intercept Blocking Buffer to cover the bottom of each well.
- Make sure to include control wells without primary antibody to serve as a source for background well intensity. Only add 50 µL of Intercept Blocking Buffer to control wells.
- Incubate with primary antibody for 2 hours with gentle shaking at RT.
Wash
-
Prepare Tween® Washing Solution as follows:
1X PBS 995 mL 20% Tween 20 5 mL 1X PBS + 0.1% Tween 20 1000 mL -
Remove primary antibody solution.
-
Using a multi-channel pipettor, add 200 µL Tween Washing Solution (RT). Add solution down the sides of the wells carefully to avoid detaching the cells from the well bottom.
-
Allow wash to shake on plate shaker for 5 minutes at RT.
-
Repeat washing steps 4 more times.
Secondary Antibodies
Dilute Secondary Antibodies
-
Dilute the fluorescently-labeled secondary antibodies in Intercept® Blocking Buffer as specified below. To lower background, add Tween 20 to the diluted antibody to a final concentration of 0.2%. Recommended dilution range is 1:200 – 1:1,200.
Goat anti-Mouse IRDye® 680RD (1:800 dilution in the combined solution)
Goat anti-Rabbit IRDye 800CW (1:800 dilution in the combined solution)
Minimize exposure of the antibody vials to light.
-
Mix the antibody solutions and add 50 µL of the secondary antibody solution to each well.
Incubate with Secondary Antibodies
- Incubate for 60 minutes with gentle shaking at RT. Protect plate from light during incubation.
Wash
- Remove secondary antibody solution.
- Using a multi-channel pipettor, add 200 µL of Tween Washing Solution at RT (Wash). Add solution down the sides of the wells carefully to avoid detaching the cells from the well bottom.
- Allow wash to incubate on a plate shaker for 5 minutes at RT.
- Repeat washing steps 4 more times. Protect plate from light during washing.
Image
- After final wash, remove wash solution completely from wells. Turn the plate upside down and tap or blot gently on paper towels to remove traces of wash buffer. For best results, scan plate immediately; plates may also be stored at 4 °C for several weeks (sealed and protected from light).
- Before plate scanning, clean the bottom plate surface and the Odyssey® Imager scanning bed (if applicable) with moist, lint-free tissue to avoid any obstructions during scanning.
- Scan plate with detection in both 700 and 800 nm channels.
Suggested Scan Settings
All settings may require adjustment for optimal data quality. Higher resolutions or scan qualities can be used, but the scan time will increase.
| Instrument | Resolution | Scan Quality | Intensity Setting (700 nm) | Intensity Setting (800 nm) |
| Odyssey Classic | 169 µM | lowest | 5 | 5 |
| Odyssey DLx | 169 µM | lowest | Auto Mode | Auto Mode |
Experimental Results

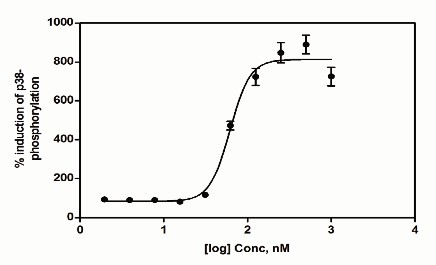
References
1. Chen, H., Kovar, J., Sissons, S., Cox, K., Matter, W., Chadwell, F., Luan, P., Vlahos, C. J., Schutz-Geschwender, A., and Olive, D. M. (2005) A cell-based immunocytochemical assay for monitoring kinase signaling pathways and drug efficacy. Analytical biochemistry 338, 136-142
2. Aguilar, H. N., Zielnik, B., Tracey, C. N., and Mitchell, B. F. (2010) Quantification of rapid Myosin regulatory light chain phosphorylation using high-throughput in-cell Western assays: comparison to Western immunoblots. PLoS One 5, e9965
3. Wong, S. K. (2004) A 384-well cell-based phospho-ERK assay for dopamine D2 and D3 receptors. Analytical biochemistry 333, 265-272
4. Kumar, N., Afeyan, R., Kim, H. D., and Lauffenburger, D. A. (2008) Multipathway model enables prediction of kinase inhibitor cross-talk effects on migration of Her2-overexpressing mammary epithelial cells. Mol Pharmacol 73, 1668-1678
5. Hannoush, R. N. (2008) Kinetics of Wnt-driven beta-catenin stabilization revealed by quantitative and temporal imaging. PLoS One 3, e3498
6. Chen, W. W., Schoeberl, B., Jasper, P. J., Niepel, M., Nielsen, U. B., Lauffenburger, D. A., and Sorger, P. K. (2009) Input-output behavior of ErbB signaling pathways as revealed by a mass action model trained against dynamic data. Mol Syst Biol 5, 239
7. Counihan, N. A., Daniel, L. M., Chojnacki, J., and Anderson, D. A. (2006) Infrared fluorescent immunofocus assay (IR-FIFA) for the quantitation of non-cytopathic and minimally cytopathic viruses. J Virol Methods 133, 62-69
8. Lin, Y. C., Li, J., Irwin, C. R., Jenkins, H., DeLange, L., and Evans, D. H. (2008) Vaccinia virus DNA ligase recruits cellular topoisomerase II to sites of viral replication and assembly. J Virol 82, 5922-5932
9. Weldon, S. K., Mischnick, S. L., Urlacher, T. M., and Ambroz, K. L. (2010) Quantitation of virus using laser-based scanning of near-infrared fluorophores replaces manual plate reading in a virus titration assay. J Virol Methods 168, 57-62
10. Lopez, T., Silva-Ayala, D., Lopez, S., and Arias, C. F. (2012) Methods suitable for high-throughput screening of siRNAs and other chemical compounds with the potential to inhibit rotavirus replication. J Virol Methods 179, 242-249
11. Wan, Y., Zhou, Z., Yang, Y., Wang, J., and Hung, T. (2010) Application of an In-Cell Western assay for measurement of influenza A virus replication. J Virol Methods 169, 359-364
12. Jamin, E. L., Riu, A., Douki, T., Debrauwer, L., Cravedi, J. P., Zalko, D., and Audebert, M. (2013) Combined genotoxic effects of a polycyclic aromatic hydrocarbon (B(a)P) and an heterocyclic amine (PhIP) in relation to colorectal carcinogenesis. PLoS One 8, e58591
13. Khoury, L., Zalko, D., and Audebert, M. (2013) Validation of high-throughput genotoxicity assay screening using gammaH2AX in-cell western assay on HepG2 cells. Environ Mol Mutagen 54, 737-746
14. Godin-Heymann, N., Ulkus, L., Brannigan, B. W., McDermott, U., Lamb, J., Maheswaran, S., Settleman, J., and Haber, D. A. (2008) The T790M "gatekeeper" mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol Cancer Ther 7, 874-879
15. Du, Y., Danjo, K., Robinson, P. A., and Crabtree, J. E. (2007) In-Cell Western analysis of Helicobacter pylori-induced phosphorylation of extracellular-signal related kinase via the transactivation of the epidermal growth factor receptor. Microbes Infect 9, 838-846
16. McInerney, M. P., Pan, Y., Short, J. L., and Nicolazzo, J. A. (2017) Development and Validation of an In-Cell Western for Quantifying P-Glycoprotein Expression in Human Brain Microvascular Endothelial (hCMEC/D3) Cells. J Pharm Sci 106, 2614-2624
17. Urlacher T, Xing K, Cheung L et al (2013) Glycoprotein applications using near-infrared detection. Poster presentation, Experimental Biology
18. Guo, K., Shelat, A. A., Guy, R. K., and Kastan, M. B. (2014) Development of a cell-based, high-throughput screening assay for ATM kinase inhibitors. J Biomol Screen 19, 538-546
19. Hoffman, G. R., Moerke, N. J., Hsia, M., Shamu, C. E., and Blenis, J. (2010) A high-throughput, cell-based screening method for siRNA and small molecule inhibitors of mTORC1 signaling using the In Cell Western technique. Assay Drug Dev Technol 8, 186-199
20. Schnaiter, S., Furst, B., Neu, J., Waczek, F., Orfi, L., Keri, G., Huber, L. A., and Wunderlich, W. (2014) Screening for MAPK modulators using an in-cell western assay. Methods Mol Biol 1120, 121-129
21. Daftarian, M. P., Vosoughi, A., and Lemmon, V. (2014) Gene-based vaccination and screening methods to develop monoclonal antibodies. Methods Mol Biol 1121, 337-346
22. Elmore, S. (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35, 495-516
23. Hanahan, D., and Weinberg, R. A. (2000) The hallmarks of cancer. Cell 100, 57-70
24. Cuenda, A., and Rousseau, S. (2007) p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta 1773, 1358-1375
25. Sui, X., Kong, N., Ye, L., Han, W., Zhou, J., Zhang, Q., He, C., and Pan, H. (2014) p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett 344, 174-179
26. Curtin, J. F., and Cotter, T. G. (2002) Anisomycin activates JNK and sensitises DU 145 prostate carcinoma cells to Fas mediated apoptosis. Br J Cancer 87, 1188-1194
27. Torocsik, B., and Szeberenyi, J. (2000) Anisomycin affects both pro- and antiapoptotic mechanisms in PC12 cells. Biochem Biophys Res Commun 278, 550-556
28. Kochi, S. K., and Collier, R. J. (1993) DNA fragmentation and cytolysis in U937 cells treated with diphtheria toxin or other inhibitors of protein synthesis. Exp Cell Res 208, 296-302
29. Kim, M., Lee, S. J., Shin, S., Park, K. S., Park, S. Y., and Lee, C. H. (2018) Novel natural killer cell-mediated cancer immunotherapeutic activity of anisomycin against hepatocellular carcinoma cells. Sci Rep 8, 10668
30. Cao, C., Yu, H., Wu, F., Qi, H., and He, J. (2017) Antibiotic anisomycin induces cell cycle arrest and apoptosis through inhibiting mitochondrial biogenesis in osteosarcoma. J Bioenerg Biomembr 49, 437-443
31. Chen, Q., Liu, X., Xu, L., Wang, Y., Wang, S., Li, Q., Huang, Y., and Liu, T. (2016) Long non-coding RNA BACE1-AS is a novel target for anisomycin-mediated suppression of ovarian cancer stem cell proliferation and invasion. Oncol Rep 35, 1916-1924
32. Liu, Y., Ge, J., Li, Q., Gu, L., Guo, X., Ma, Z. G., and Zhu, Y. P. (2013) Anisomycin induces apoptosis of glucocorticoid resistant acute lymphoblastic leukemia CEM-C1 cells via activation of mitogen-activated protein kinases p38 and JNK. Neoplasma 60, 101-110
33. Stadheim, T. A., and Kucera, G. L. (2002) c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for mitoxantrone- and anisomycin-induced apoptosis in HL-60 cells. Leuk Res 26, 55-65